Our Research
Here are some (but not all) of our research interests. For more information, please visit the publications page.

Contact Cluster Modeling of Allosteric Communication in PDZ Domains

Combining single molecule FRET measurements and MD simulations to elucidate hierachical dynamics in allostery
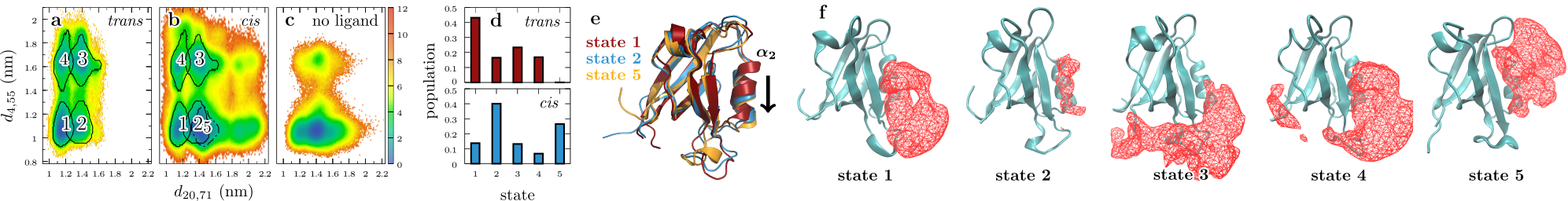
Real-time observation of ligand-induced allosteric transitions
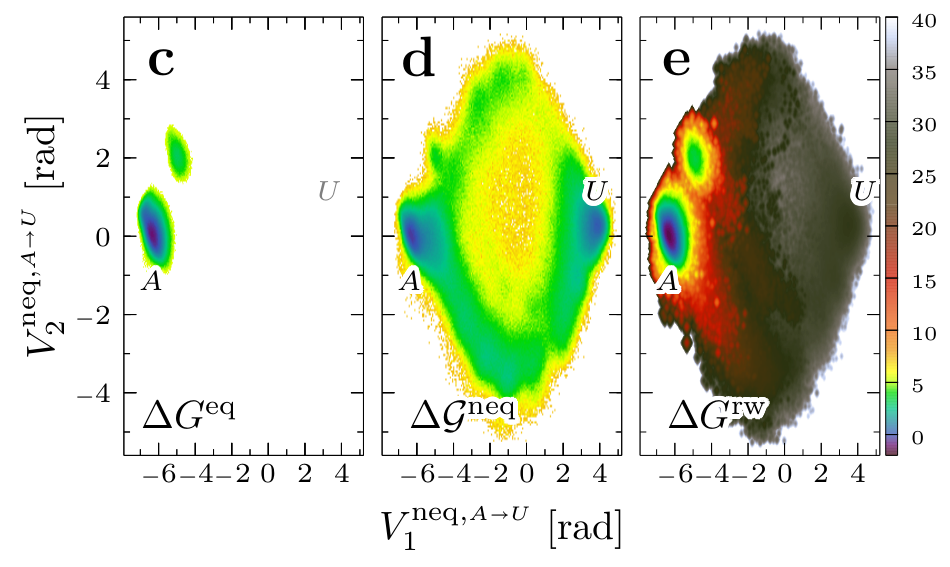
Nonequilibrium MD simulations of a proteins allosteric communication
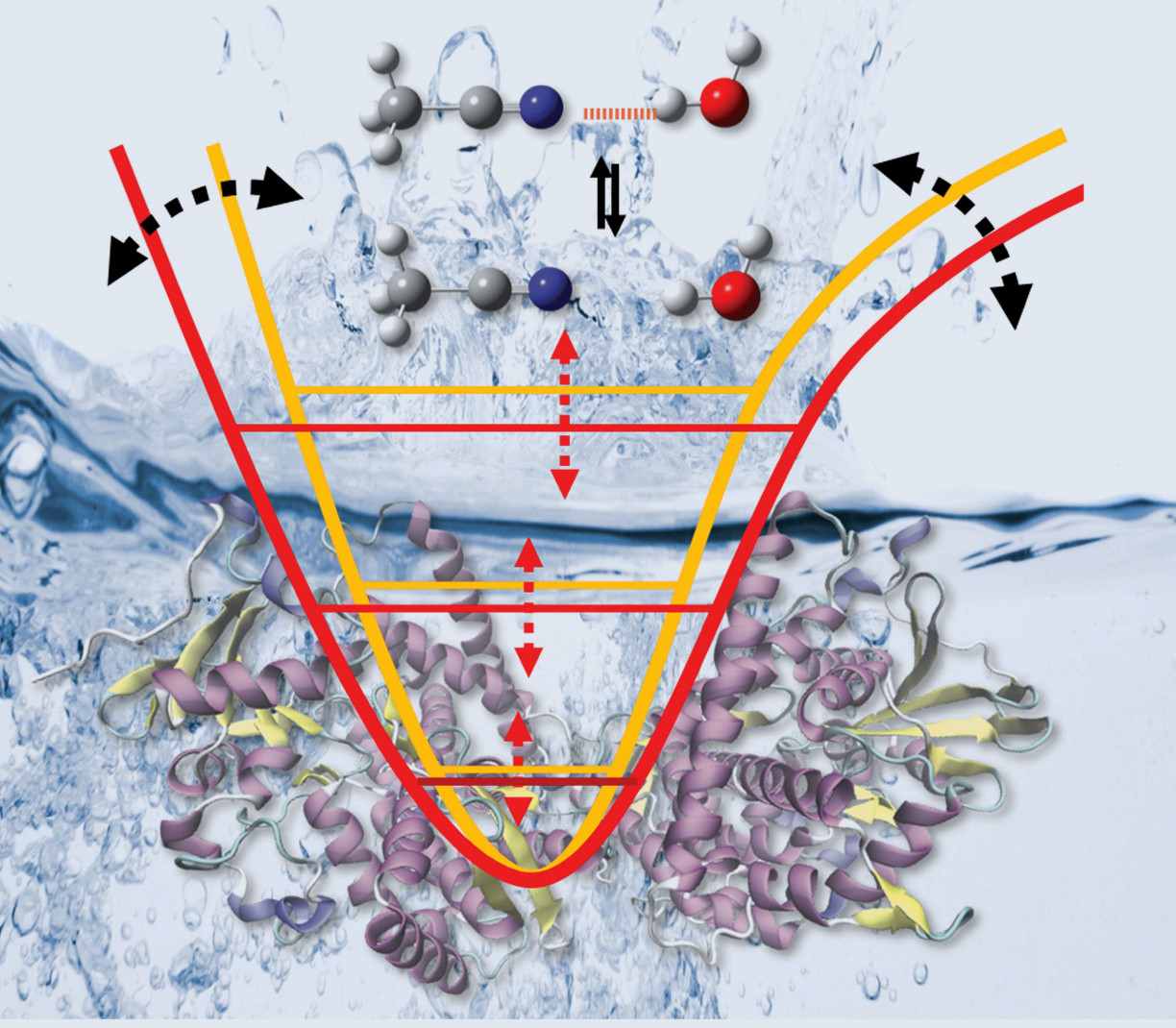
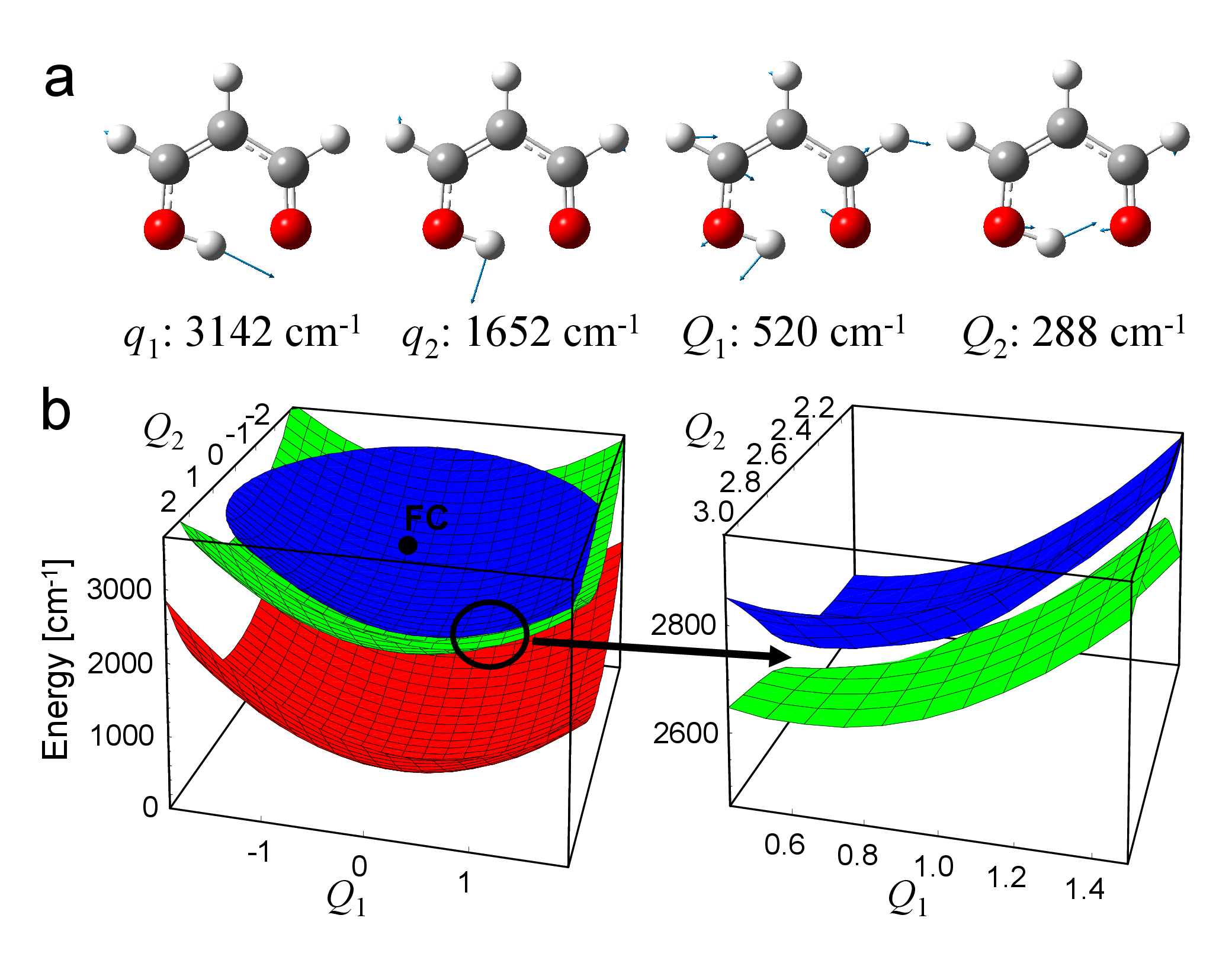
- the femtosecond vibrational energy redistribution from localized high-frequency modes to delocalized low-frequency modes,
- transport of energy through the molecule, and
- the cooling the molecules due to heat transfer to the solvent.
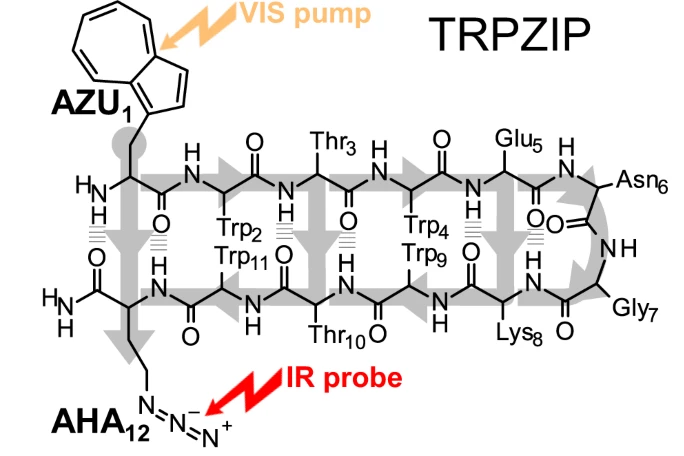
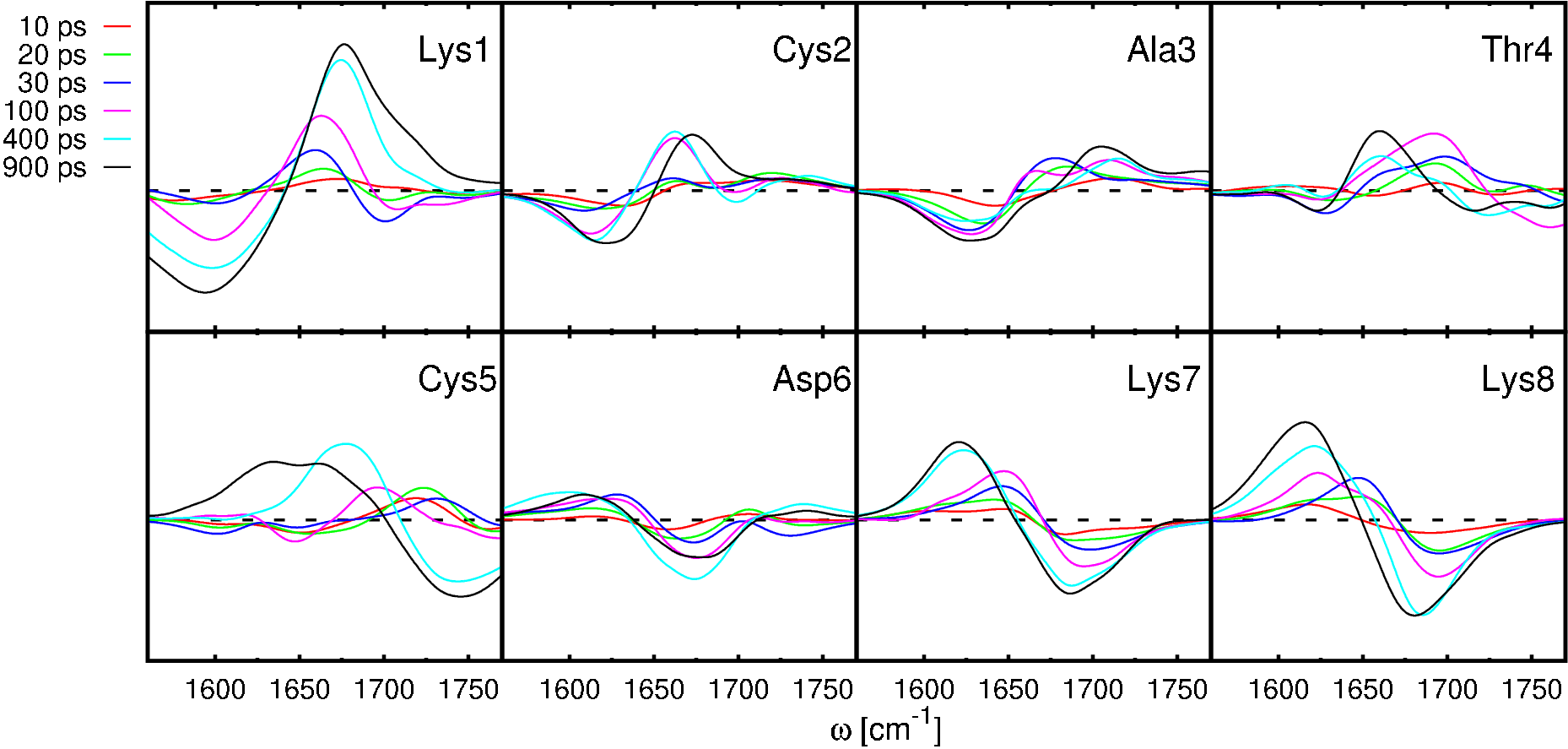
Through Bonds or Contacts? Mapping Protein Vibrational Energy Transfer Using Non-canonical Amino Acids
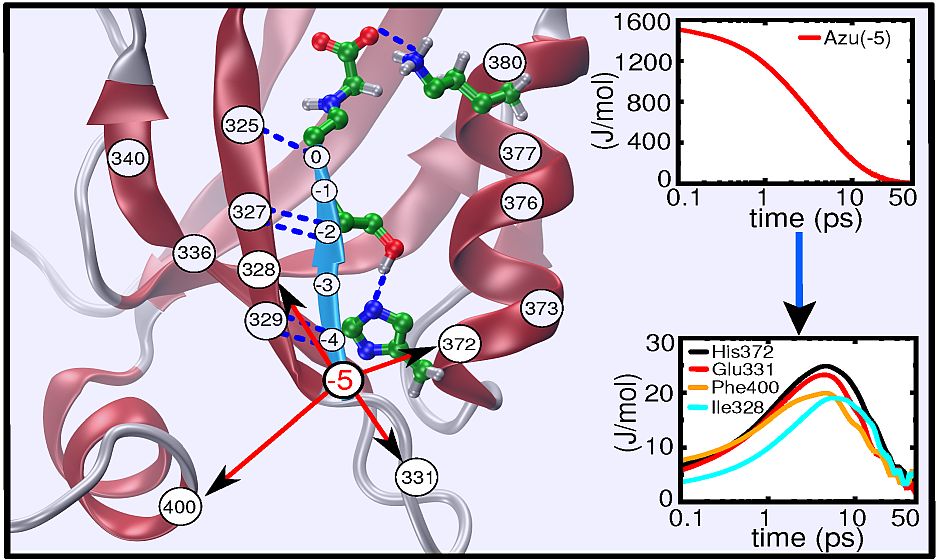
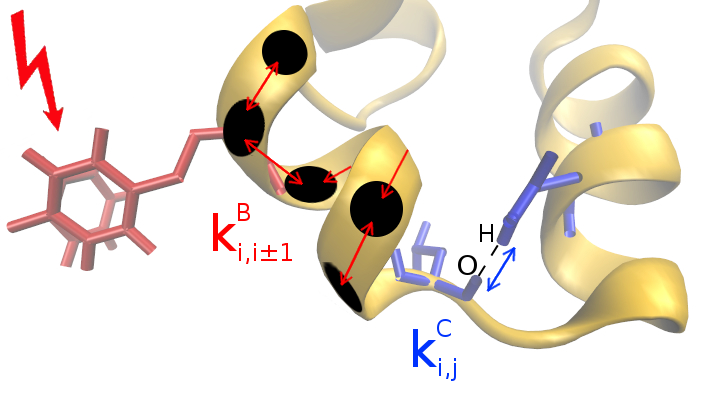
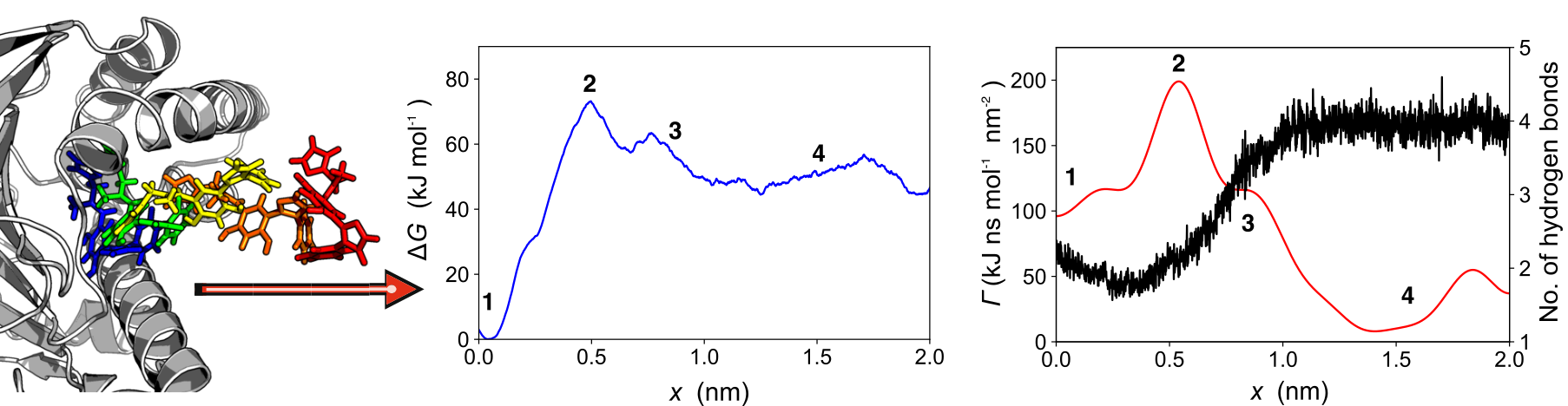
Scaling Rules for Energy Transport
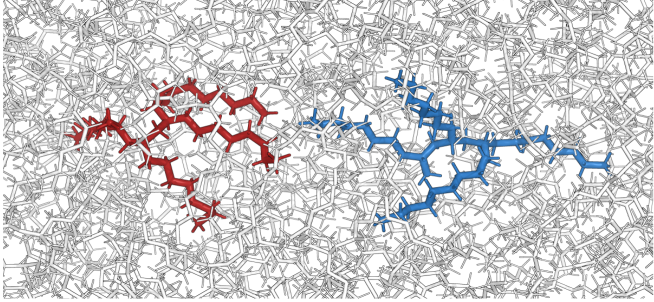
Molecular Origin of Driving-Dependent Friction in Fluids

Log-periodic oscillations as real-time signatures of hierarchical dynamics in proteins
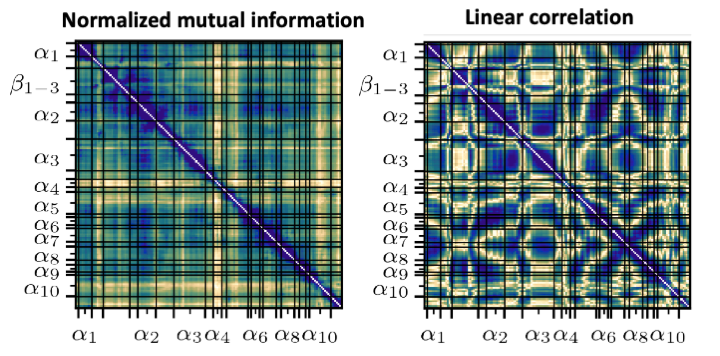
Accurate estimation of the normalized mutual information of multidimensional data

Correlation based feature selection to identify collective motion
PCA of nonequilibrium MD simulations
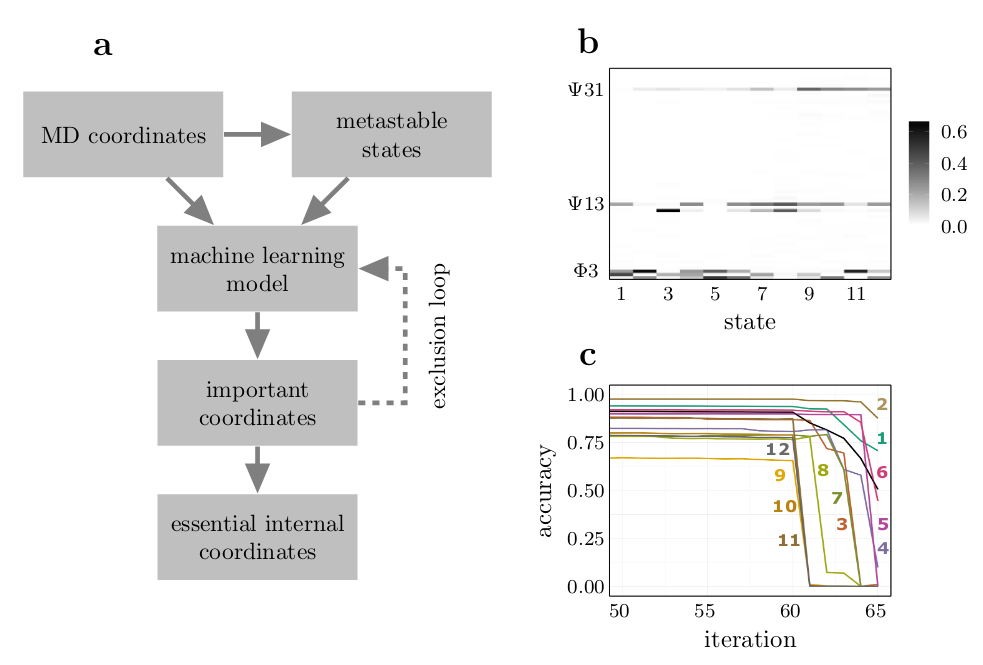
Learning the essential coordinates
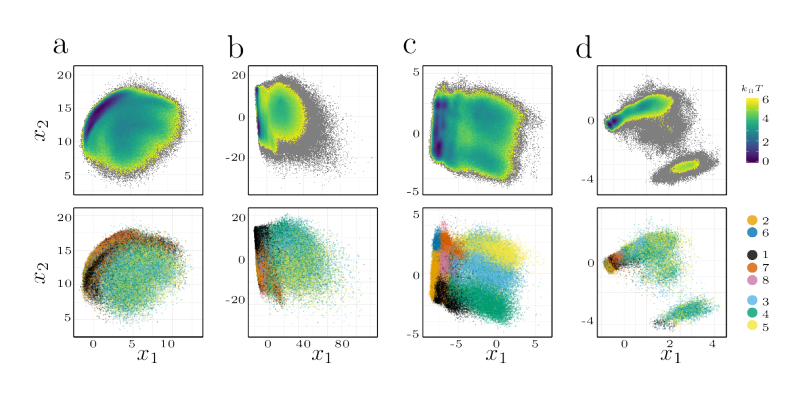
Principal component analysis - Cartesian vs. internal coordinates


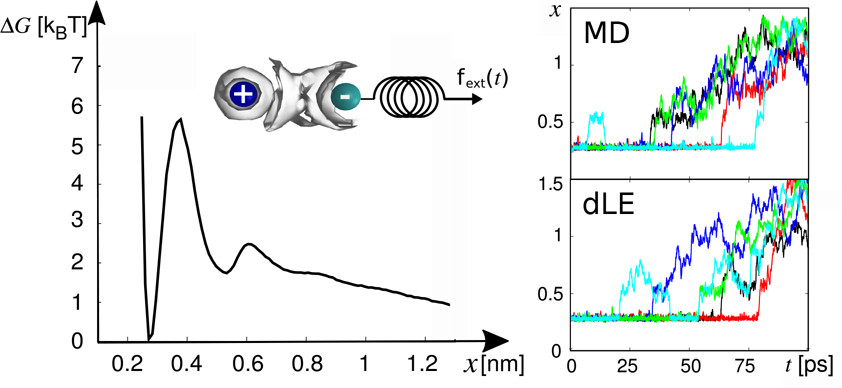
Data-driven Langevin modeling of nonequilibrium processes
Vibrational Spectroscopic Map, Vibrational Spectroscopy, and Intermolecular Interaction
Real time observation of ultrafast peptide conformational dynamics: MD simulation vs. IR experiment
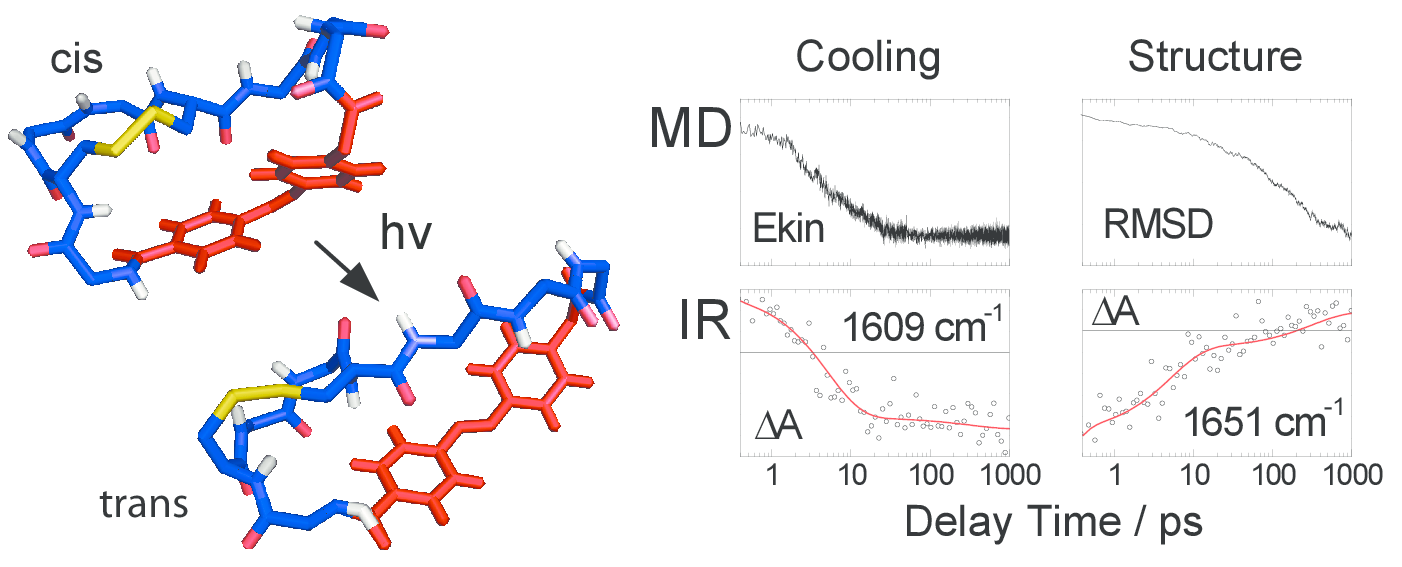
Simulation of transient infrared spectra of a photoswitchable peptide
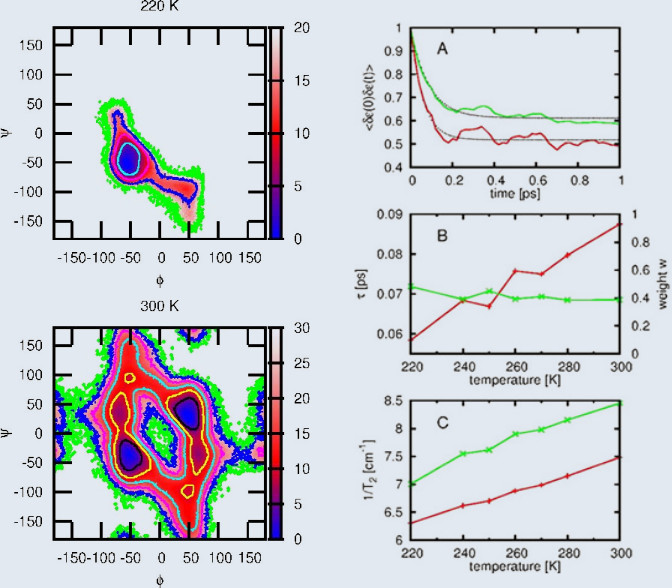
Right: (A) Short-time evolution of the total C=O frequency fluctuation correlation function at 220 K (red) and 300 K (green). The curves are fitted by an exponential function, the weight (green) and decay time τ (red) of which are shown in panel (B) as a function of temperature. The resulting total homogeneous dephasing rate (red) is shown in (C) together with the antidiagonal width of the 2D-IR spectra (green).
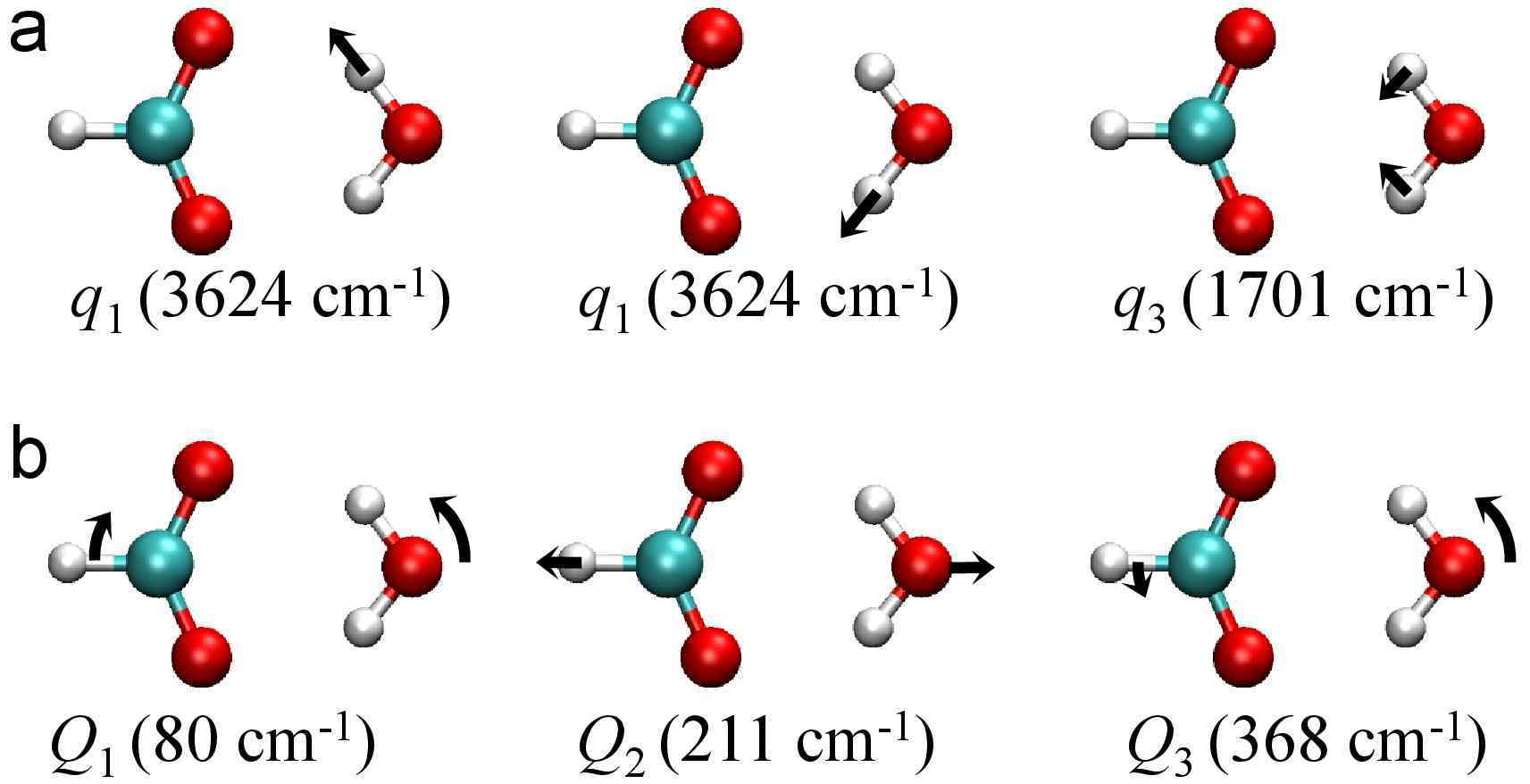
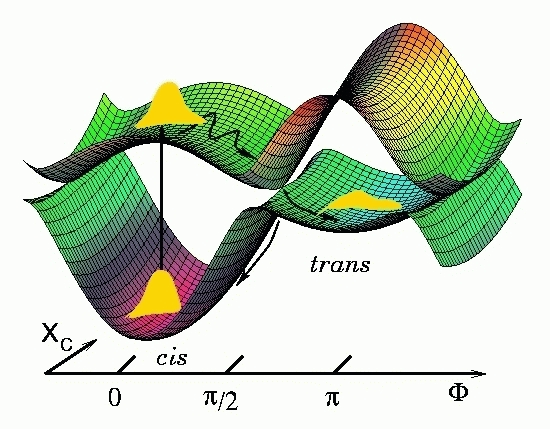
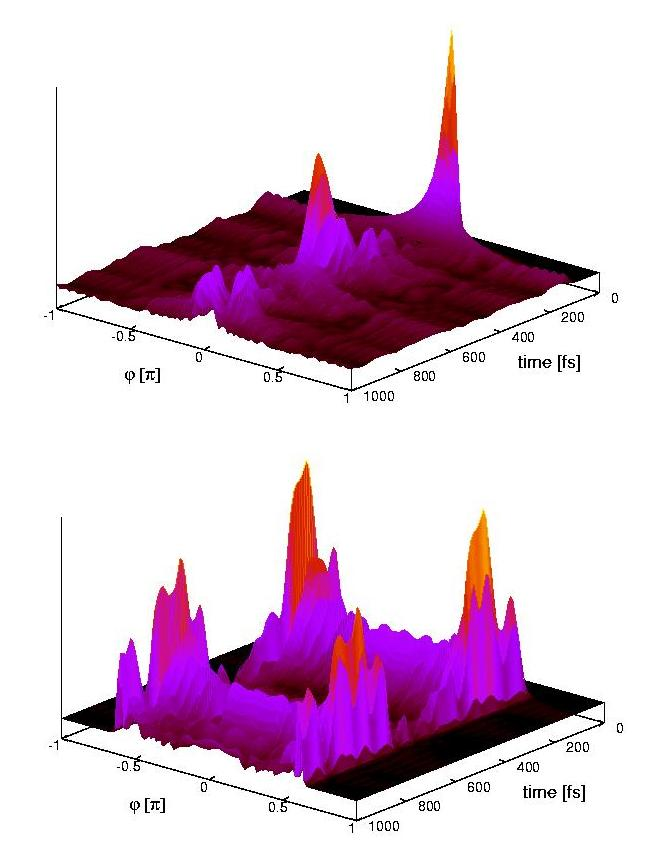
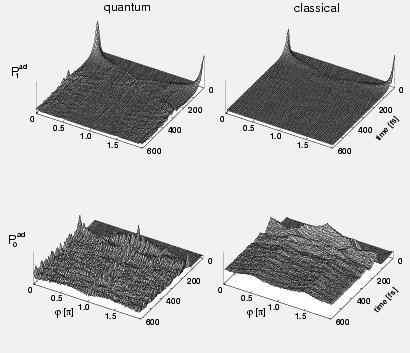
Ultrafast nonadiabatic photoreactions
Maximum Caliber
- It is partition-function-based, so we can draw insights from similarities to equilibrium statistical mechanics,
- it is trajectory-based, so it gives more dynamical information than population-based approaches like master equations; this is particularly important for few-particle and single-molecule systems,
- it gives an unambiguous way to relate flows to forces, which has traditionally posed challenges, and
- like Maximum Entropy, it may be useful for data analysis, specifically for time-dependent phenomena.